IUPs
Intrinsically unstructured/disordered proteins have no single well-defined
tertiary structure in their native state. In many cases a protein is fully
disordered, while in many other cases there are long disordered segments in
otherwise ordered, folded proteins. The structure of some IUPs resembles the
denatured states of ordered proteins, best described as an ensemble of rapidly
interconverting alternative structures, while others have some tendency to form
local secondary structure elements or show molten-globula like
characteristics. Despite their lack of a well-defined globular structure, these
proteins carry out basic functions, mostly associated with signal transduction,
cell-cycle regulation and transcription. Often, their function is realized via
molecular recognition in which structural disorder confers specific advantages,
such as increased speed of interaction and specificity without excessive
binding strength. Disordered regions can also constitute flexible linkers, or
spacers that have a role in forming macromolecular assemblies. Some well
studied examples of IUPs include p21, the N-terminal domain of p53 or the
transactivator domain of CREB. The importance of protein disorder is further
underlined by its prevalence in various proteomes. In some eukaryotic genomes
more than 20% of the coded residues are predicted as disordered.
Most protein structures known so far have been elucidated by X-ray crystallography.
However, this technique is not suitable to characterize disordered regions of
proteins, as corresponding regions are missing from the electron density map due to
their intrinsic flexibility. NMR spectroscopy is the most powerful method to obtain
site-specific information on the structure and dynamics of IUPs. Other techniques,
including CD and other spectroscopic methods, hydrodynamic measurments, proteolytic
degradation can also give important information about protein disorder. However, as
each technique probes different aspects of protein structure, they do not
necessarily correctly identify disorder. For example, loopy proteins, which have no
repetitive secondary structure, would appear disordered by CD but ordered by the
other techniques. With NMR, disorder often is concluded from poor signal dispersion,
which does not distinguish between random-coils and molten globules of high
potential to fold in the presence of a partner. In X-ray crystallography, crystal
packing may enforce certain disordered regions to become ordered, and disordered
binding segments are often crystallized in complex with their partner and are
classified ordered despite their lack of structure in isolation. In addition, wobbly
domains would appear disordered, despite their intrinsic structural order. As a
result, available datasets are rather limited in size and are heterogeneous in terms
of experimental conditions, techniques and interpretation of data. They also lack in
consistency due to the absence of clear conceptual and operational definition(s) of
structural disorder. All these result in false positive- and false negative
classifications, i.e. the inclusion of ordered segments in disorder databases and
the exclusion (and inclusion in ordered reference database) of disordered
proteins/segments. In consequence, predictors trained on these datasets for
assessing disorder reflect these uncertainties. Our method, however, was
parameterized using only globular proteins, and does not suffer from these
uncertainties.
The basis of predicting protein disorder is the difference in sequence
characteristics between folded and disordered proteins. Typically, IUPs exhibit
a strong bias in their amino acid composition and even a reduced alphabet is
able to recognize them at the level of complete sequence. Other results
indicate, however, that there are differences in sequence properties among
different types of disordered proteins. Various factors have been suggested to
be important in terms of protein disorder, including flexibility, aromatic
content, secondary structure preferences and various scales associated with
hydrophobicity. Beside low mean hydrophobicity, high net charge was also
suggested to contribute to disorder. All these different analyses, though, hint
that the amino acid composition of IUPs results in their inability to fold due
to the depletion of typically buried amino acids and enrichment of typically
exposed amino acids, which implies that globular proteins have
sequences with the potential to form a sufficiently large number of favorable
interactions, whereas IUPs do not. In this method we put this inference on a
quantitative footing by taking an energetics point of view. On this ground, the
sequences encoding for globular proteins and IUPs can be distinguished.
Short description of our method
Our prediction method for recognizing ordered and disordered regions in proteins
is based on estimating the capacity of polypeptides to form stabilizing contacts. The
underlying assumption is that globular proteins make a large number of interresidue
interactions, providing the stabilizing energy to overcome the entropy loss during
folding. In contrast, IUPs have special sequences that do not have the capacity to
form sufficient interresidue interactions. Taking a set of globular proteins with
known structure, we have developed a simple formalism that allows the estimation of
the pairwise interaction energies of these proteins. It uses a quadratic expression in
the amino acid composition, which takes into account that the contribution of an amino
acid to order/disorder depends not only its own chemical type, but also on its
sequential environment, including its potential interaction partners. Applying this
calculation for IUP sequences, their estimated energies are clearly shifted towards
less favorable energies compared to globular proteins, enabling the predicion of
protein disorder on this ground.
The calculation involves a 20 by 20 energy predictor matrix. The parameters of
this matrix were derived using globular proteins with known structures only.
For these structures, the energy can be calculated using a coarse-grained
approach, as a sum of pairwise interactions between amino acid pairs within a
distance cutoff. The applied interaction matrix was calculated from the
observed frequencies of amino acid pairs using the approach of Thomas and Dill
(PNAS 1996, 93, 11628-11633). We aimed to estimate the pairwise energy
without a presumed structure, from the amino acid composition only. The
parameters of the energy predictor matrix were determined by least squares
fitting in a way to obtain the best fit between the energy
calculated from the known structures and the energy estimated from the amino
acid compositions for all proteins in the dataset overall.
This novel approach is validated by the good correlation of this estimated
energy with the values calculated for known structures. When applied for
disordered sequences, their predicted energy values was clearly shifted towards
less favourable energies compared to globular proteins. This indicates that
experimentally characterized disordered proteins have special amino acid
compositions, which, independently of the actual sequence, do not allow the
formation of favorable contacts expected for folded proteins. Thus, these
proteins are indeed intrinsically unstructured.
Based on the significant separation between the estimated pairwise energies
of globular and experimentally verified intrinsically unstructured proteins,
this approach can be turned into a method to predict protein disorder. Since
many proteins are not fully ordered or disordered, we considered the local
sequential neighborhood only for this purpose. The original energy predictor
matrix P, derived at the level of global sequences, was recalculated by
treating each position separately, and taking into account only its predefined
neighbourhood in sequence. The energy and amino acid composition for each
position was calculated only by considering interaction partners 2 to 100
residues apart. The choice of this range represents a trade-off between the
intention of covering most structured domains, but separating distinct domains
in multi-domain proteins. This procedure yields an estimated energy at position
p of type i:
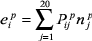
where Pp is the position specific energy predictor matrix.
The position-specific
estimation of energies were averaged over a window of 21 residues.
|
